12 向疾病挑戰:防治遺傳疾病
歷經努力,在37歲第一次懷孕后,凱瑟琳·麥克歐利福(Kathleen McAuliffe)做了羊膜穿刺術,並放心地得知她的胎兒只有兩條第21號染色體。但她不知道的是,從這個檢測也可以看出其他的染色體異常。細胞遺傳學家發現她胎兒的第2號染色體有倒位的情況:這就像這條染色體上有一個區段跑出來,翻了個身後,以跟原先相反的方向再嵌回原來的染色體上。但是除了這個消息以外,他們無法提供任何其他有用的建議,這個倒位有可能會造成問題,例如它可能造成遺傳物質不平衡,但是它也可能完全沒有作用。若要找到更多的信息,就得檢查麥克歐利福夫婦的第2號染色體。如果他們當中任一人也有倒位情形(換句話說,這並不是胎兒的自發性變化),就可以推論這個倒位沒有或只有極少的影響,畢竟雙親都是正常的。但是麥氏夫婦兩人的第2號染色體都沒有倒位現象,意味著這是在精|子或卵子內初次發生的。這種倒位現象對胎兒有什麼影響?凱瑟琳發現自己必須作一個攸關生死的決定。
這個問題不但攸關個人,確實也攸關社會。X染色體脆弱症的「前突變」發生率高(可能高達每200個X染色體中,就有1個有這種前突變),可以作為進行族群篩檢的正當理由。在美國,根據估計,一個重症病人一生不工作並待在收容所里,大約要花掉200萬美元。由於要提供負擔得起的照顧愈來愈難,因此這一點成為提供每位母親檢測機會的有力論點。
根據各方說法,這個事件的主角是克萊恩(Martin Cline),一個聰明且雄心壯志的臨床醫生,向來致力於減輕病人的痛苦。他對β-地中海型貧血症特別感興趣,也就是摩戴爾(Bernadette Modell)在倫敦的塞普勒斯族群中篩檢出的血紅素疾病。在動物實驗成功后,克萊恩向他任職的加州大學洛杉磯分校審查委員會,申請使用非重組DNA (nonrecombinant DNA)進行基因療法的人體實驗。當他的申請仍在審查過程中時,過度熱衷的克萊恩卻已經安排對兩名非美國境內的婦女(分別在以色列和義大利)進行實驗,但他使用的是重組基因,當時國家衛生研究院的規定仍禁止使用重組基因。在回到洛杉磯后,克萊恩發現他的申請被駁回;審查委員會裁定必須要有更多動物實驗的數據,才能批准人體實驗。克萊恩幾乎違反了所有的規定:他不僅在沒有獲得授權下就治療人類,還使用了明確被禁止的方法。克萊恩也為此承受了苦果,他失去聯邦經費,並被迫辭去系主任的職位。基因療法也失去了它的第一個醫生。
杜克大學(Duke University)的羅塞斯(Allen Roses)沒有跟著大夥走,反而選擇研究常見得多的晚髮型阿爾茨海默氏症。晚髮型僅偶爾出現在家族中,例如,在1984年宣布患病的前總統里根,他在兩年後失去了死於同一型阿爾茨海默氏症的哥哥尼爾(Neil),他們的母親也是死於這一型。
這名少年的病逝不僅對他的家人是一大打擊,也令研究圈震驚。詳盡的調查發現,這當中有嚴重的程序缺失,最顯著的錯誤是:雖然先前在相同的研究中已有兩名病患出現肝中毒的癥狀,但這些病例卻沒有通報給任何管制當局,也沒有告知自願參与研究的患者。如果蓋辛格家知道這點,他或許不會這麼急著自告奮勇,說不定他至今仍活著。這場悲劇對基因療法的進展是一大打擊。有一陣子,美國食品及藥物管理局要求全美國的大學及其他數個研究計劃終止這類實驗。參議院惟一的醫生,來自田納西州的佛瑞斯特(Bill Frist)也深入調查了人體實驗的通報程序;柯林頓總統則要求改進「告知同意」的標準,認為實驗對象有權獲知所有的潛在風險。如果蓋辛格的事件帶來任何正面效果,那就是聯邦政府對人體實驗的監督愈來愈嚴格。
痛苦許久后,凱瑟琳認為這種不確定性太大,決定終止懷孕。儘管她特別要求不要把解剖結果通知她,畢竟她仍為失去這個胎兒感到哀傷又充滿罪惡感,但是在行政疏失下,解剖報告仍送達她家,而她發現胎兒真的有嚴重異常的情況。但這並不值得欣慰,至今她仍把胎兒的超音波影像收在抽屜里。幸好她後來再度懷孕,而且胎兒都沒有任何問題。現在她是個幸福的媽媽,有兩個小孩,而且如她所說,「他們健康得吵死人」。
1990年9月,阿善蒂率先接受了這個療法;辛迪則是在4個月後。她們每幾個月就注射一次經過基因改良的免疫細胞。同時,她們仍繼續接受與基因無關的酶替代療法,跟高雪氏症患者的療法相同,但是劑量較少。這是美國國家衛生研究院的人類基因療法小組委員會要求的預防措施,他們認為在沒有任何安全保障的情況下讓這兩個女孩接受新療法太過危險,這論點很合理。雖然這個實驗是在不完全控制的情況下進行的,但似乎仍獲得成效:兩個女孩的免疫系統都有改善,而克服輕微感染的能力也變得較好。我個人可以作證,辛迪在1992年和家人造訪冷泉港實驗室時,看起來就像個非常健康的11歲女孩。然而在10餘年後,這個實驗仍無法獲得完全確定的結果。現在阿善蒂的免疫系統功能正朝正常的水平前進,但她的T細胞(胸腺淋巴細胞)大約只有1/4是來自基因療法。在辛迪的血液中,來自基因療法的T細胞所佔的比例更小,但現在她的免疫系統運作得很好。不過我們還是很難說這兩個女孩的進展有多少是基因療法的成果,又有多少是持續接受酶療法的成果。這樣的結果太過模糊,所以不能毫無保留地算是基因療法的成功。
有些人反對擴大篩檢範圍,是基於比較抽象的原因。有些人認為篩檢等於自承失敗,是一種錯誤的解決方式。支持團體的宗旨就是要讓患者有社群歸屬感,覺得受到社會的重視。而篩檢所促成的結果,直截了當地說,就是墮掉染病的胎兒,這不就和這些支持團體的宗旨相互衝突了嗎?
第一個顯然獲得成功的基因療法,是1990年美國國家衛生研究院的安德森(French Anderson)、布萊斯(Michael Blaese)和卡爾弗(Ken Culver)進行的。他們選擇治療的目標是一種非常罕見的疾病,稱為腺苷脫氨酶(adenosine deaminase, ADA)缺乏症,病患的免疫系統因為缺乏一種酶而喪失功能,造成跟「泡泡男孩」維特一樣無法抵抗疾病。他們的實驗對象是兩個小女孩,4歲的阿善蒂·狄西瓦(Ashanti DeSilva)以及9歲的辛迪·柯特薛爾(Cindy Cutshall)。
有時這些影響不是發生在當下這一代身上,而是在未來的世代身上。X染色體脆弱症(fragile X)是遺傳性心智遲緩最常見的形式。(唐氏症更常見,但為自發性,通常不是經由遺傳得到。)除了智商低以外,X染色體脆弱症患者的典型癥狀是臉形特別長,下巴和耳朵特別大,極度好動,偶爾會有易怒的性情。這種病跟假肥大型肌營養不良症一樣,都是性聯遺傳(導致這種病的基因位於X性染色體上),不同的是,男女都有可能得病。擁有這個基因的一個正常備份,顯然不足以壓抑突變基因的影響;但女性的癥狀通常比較輕微,而且女性的得病幾率是1/8300,男性則是1/5000。造成X染色體脆弱症的突變,跟造成亨廷頓氏症的突變類似:它是因DNA三聯體CGG一再重複而引起的。正常人的CGG重複數約為30個,但X染色體脆弱症患者的CGG重複數至少有50個,有時甚至多達90個。由於我們目前尚未完全明白的原因,CGG重複數一般會隨著每個世代而增加;一旦有大約230個CGG三聯體,這個基因將無法再製造信使RNA,從而無法再發揮功能。這種病的名稱來由是因為這些重複會使X染色體的結構明顯弱化,易於斷裂。
2000年,《美國人類遺傳學期刊》(American Journal of Human Genetics)發表了一項調查結果,這項調查詢問健康保險業者,如果可以獲得遺傳信息的話,他們是否會根據遺傳信息來調整保費的費率?他們會不會打算要求健康狀況良好,但因基因突變而容易患病的客戶付更高的費用?大約有2/3的受訪者坦承會,而剩下的1/3十之八九在說謊。保險公司不是慈善機構,而是生意人,必須讓股東滿意。我們沒有理由假定,他們在可以自行其是的情況下會改變向來的做法,亦即把有風險的客戶保費提到最高,而盡量避免替最可能申請理賠的客戶承保。這份報告也引述了一個案例:有一家保險公司懷疑一位客戶患有遺傳疾病而提高其保費費率,只因為這人曾經申請做亨廷頓氏症的診斷檢測。
就在基因療法界仍因蓋辛格死亡事件的衝擊而震驚不已時,法國傳來令人振奮的成功消息。法方鎖定的目標是嚴重綜合型免疫缺乏症(SCID),亦即造成維特終生以「泡泡」為家的疾病。雖然骨髓移植可以達到治療效果,但成功率只有40%左右,即使移植成功,也經常有可怕的併發症,如同維特不幸的案例。2000年,費希爾(Alain Fischer)在巴黎內克醫院(Necker Hospital)領導的醫療小組,對兩名嬰兒進行了基因療法,他們跟維特一樣,也是一出生就住在無菌的隔離空間里。這個小組採取跟治療腺苷脫氨酶缺乏症相同的方法,以反轉錄病毒為載體,把需要的基因送入取自嬰兒的細胞,再把這些細胞重新注入嬰兒體內。但法國小組作出了一個相當創新的改變,他們是從嬰兒的骨髓中取得的待改造細胞。這個方法運用的是骨髓的免疫幹細胞,而不是一般在血液中找到的普通T細胞,如果成功的話,這個方法可以成為自發永續的基因療法。幹細胞在複製時,不僅本身的數目會增加,由它們自然分化形成的特化體細胞(specialized somatic cell)也會增多。因此,由經過改造的幹細胞所產生的T細胞,也會攜帶相同的新插入基因,患者就不必持續注射改造過的細胞。
戴維·維特(David Vetter)自從出生之後就沒有直接接觸過另一個人類,因為他罹患了嚴重綜合型免疫缺乏症(severe combined immunodeficiency disorder, SCID)。他的身體無法製造B細胞和T細胞,由於這兩種細胞都是免疫系統對疾病產生反應的重要成分,所以他只要輕微感染就會生病。
戴維的雙親在他出生前就知道他可能罹患SCID,他們的第一個兒子就是死於這種病。這次維特夫婦和醫生們都已有所準備,他們很早就決定,如果這個嬰兒證明罹患SCID,他會在無菌的隔離環境里生活,直到治療SCID的方法問世,以醫學進展的速度來看,這應該不會太久。戴維在1971年9月以剖腹產的方式來到人世,而且立即被安置在一個無菌保育器里。任何人跟他接觸時,都必須使用內建於這個小保育器里的乳膠手套。隨著他逐漸長大,他也被移至愈來愈大的無菌環境,亦即塑料「泡泡」里,但是有一樣東西一直沒變:乳膠手套。它們是他觸摸外界任何人或物體的惟一方式。
以這類方法來解決遺傳缺陷所造成的禍害,顯然是很合理的做法,但是專業人士或一般民眾對基因療法的構想,卻不是很能接受。其實這種反應也不足為奇:一個連對穀物進行基因改造都極其謹慎的文化,想當然會反對基因克隆人類(你也可以稱之為基因改造人類[GM human]),無論其潛在好處有多大。這樣的文化會對生殖細胞療法更加反彈也是可以預期的,因為操控DNA有破壞基因的危險。在體細胞基因療法中,損壞造成的效應可能有限;但在生殖細胞療法中,這類損壞有可能意外造成殘障。即便是支持這些療法的人,包括我在內,也會在確保這種技術絕不會造成意外傷害后,才會建議實行。不過,許多科學家堅信我們永遠不該嘗試種系基因療法。無論是基於道德觀或對未知事物毫無緣由的恐懼,我認為這類論點終究無法令人信服。基本上,生殖細胞療法只是把無意間出錯的事物矯正回來。但是目前這在學術上仍有爭議的原因是,生殖細胞療法仍然遠遠超出我們現有的技術能力。在能掌握它之前,我們應該把重心放在發展體細胞基因療法上,使其成為一個強大的工具。
於1989年首度施行的胚胎著床前檢驗,目的在於測知胚胎的性別;如果有可能罹患的是性聯遺傳疾病,例如假肥大型肌營養不良症,那麼性別是相當重要的信息。一名是帶因者的母親可以選擇只懷女胎,因為女孩雖然有可能是帶因者,但是不會發病。後來把胚胎著床前遺傳診斷術的用途,從簡單的
九*九*藏*書性別鑒定擴大至偵測特定突變的人,是溫斯頓的同事漢迪塞德(Alan Handyside)等人;1992年,他們首度用這個技術來檢測不是性聯遺傳的纖維囊泡症。
副作用向來是醫學難以擺脫的問題。藥物可能不只對原先鎖定的目標造成影響,而手術則可能會引起併發症。雖然在許多方面,基因療法都與傳統醫學不同,但現在我們已經知道它仍擺脫不了發生意外結果的定律。費希爾的SCID療法可能在修正原有基因的過程中,無意間產生了新的問題。畢竟,任何把病毒DNA插入病人細胞DNA的療法,原本就潛藏著風險,因為這個外來DNA有可能偶然地阻斷某個重要基因的功能。由於發生這種情況的細胞通常都會死亡,因此這樣的意外一般不會造成影響。但是這個遭阻斷的基因在喪失功能后,其後果有可能不是導致細胞死亡,而是使細胞毫無節制地增殖;這也就是說,病毒基因插入法有可能引起癌症。這似乎就是那名SCID寶寶發生的情況。
儘管社會上存有抗拒心理,讓大規模遺傳篩檢的好處無法發揮,令人沮喪,但在短暫的篩檢史上,也並不完全都是小規模的實驗性研究,以及一片撻伐之聲。還是有一些快樂和發人深省的故事,說的是對高危族群進行遺傳疾病篩檢的成功案例。
儘管個人的權利不會因為DNA所揭露的信息而受到侵害,但在心靈上可能無法這麼容易恢復平靜;如同南西·魏克斯勒深切體會到的,基因認知可能會揭露可怕的未來。我同意她的看法:知道一些我們無力補救或改善的事情,一點意義也沒有。阿爾茨海默氏症是我這個年紀的人最擔憂的事情之一,但是在缺乏經過驗證的療法下,我一點也不想檢驗我是否有APOEε4等位基因。溫特確定擁有一份APOEε4,我們之所以知道,是因為他堅持公開宣布賽雷拉基因公司所定序的基因組是他的。儘管這個等位基因在膽固醇處理過程中具有一定的作用,但它不僅會使人患阿爾茨海默氏症的幾率提高,連患心臟疾病的幾率也會提高。(在任何方面,APOEε4都不是一項有利資產。)在得知這個基因事實后,溫特明智地盡量釆取預防措施:他服用一種叫做他汀(statin)的藥物,這種葯可以降低膽固醇含量,而且或許可以阻礙或預防阿爾茨海默氏症的發生。即使我不知道自己的APOE等位基因情況,但我也在服用他汀,我的想法是反正採取一些預防措施也沒什麼不好。如果他汀真的如藥廠所宣稱的那麼有效,我們就可以多看幾年溫特引起的爭議(希望也會有沃森引起的爭議)。
在20世紀90年代早期,當時任職於密歇根大學的柯林斯跟加州大學柏克萊分校的瑪麗-克萊爾·金合作尋找BRCA1。他們採取標準做法:收集家族、準備DNA樣本、測試標誌,這一切都著眼于這個基因上。一個超過50人的家族中,有多起乳癌病例,顯然這個家族的體質容易罹患乳癌。1992年9月,這個家族的一名成員(姑且稱她為「安妮」)向柯林斯的同事韋伯(Barbara Weber)透露,她準備在下星期做雙側乳|房切除術,雖然還沒有跡象顯示她已經罹患癌症,但她決定不再忍受對未來的不確定感,而想採取這種激烈的預防手段。然而韋伯從DNA分析中獲得的結論是,安妮罹患乳癌的幾率並不特別高,事實上,她得乳癌的幾率跟沒有家族病史的婦女一樣。但是這個推論是以研究項目為背景的,而事前他們早已達成共識,這類數據不得用於臨床診斷。
如同亨廷頓氏症、假肥大型肌營養不良症和許多其他的遺傳疾病,主導X染色體脆弱症相關研究的人,也是受這種疾病影響最深的人:患者的家人與摯愛的人。X染色體脆弱症協會(簡稱FRAXA)在募集捐款與促使國會支持X染色體脆弱症研究上,成效卓著。雖然有些科學家可能認為,這類團體只不過是提供那些陷入悲慘困境的人一些虛幻的安慰,讓他們以為自己並非完全無能為力而已。經驗顯示,這些熱心奉獻、資源豐富,最重要的是動機強烈的組織,例如FRAXA,有時的確能突破萬難,是破解這些疾病的重要關鍵。對於那些在財務和科學上都投下龐大賭注的人來說,有時在命運的協助下,會獲得最大的報償。
20世紀60年代,簡單的血液檢驗發展出來以後,篩檢計劃在全美各地倉促展開。儘管立意良善,但它們造成的傷害卻比帶來的好處還多。篩檢人員一般沒有正確地告知檢驗對象有關這個篩檢及其結果的重要性。許多診斷是帶因者的人誤以為自己患有這種疾病;有些人甚至因此找不到工作或無法參加健康保險。有可能把患病危險傳給下一代的夫婦,被強力勸告必須考慮生孩子的後果。這些篩檢計劃(有些是強制性的)實質上形成一種壓制,讓一些人感到種族優生學又在美國復辟,檢驗結果呈陽性的人全都被打上了歧視的烙印。悲哀又諷刺的是,從純粹醫學觀點來看,這個篩檢運動其實是很有道理的:當時儘管在治療上已有進展,但鐮形細胞貧血症仍是痛苦的慢性病。對這種預防勝於治療的疾病,篩檢是最好的方法,但是最初設計來根絕它的機制在執行上卻有很大的缺失,反而激怒了許多原本想造福的對象。
辛迪·柯特薛爾是最早參与基因療法的病人。在到冷泉港實驗室參觀后,她給沃森寄了一張沃森演講時的圖畫。
在丹佛這項研究中,最有意思的部分或許是學生家長和監護人對海格曼提議的反應。他們大多認為這個診斷具有好處,可以改善孩子的教育,也能檢驗出家族血統中是否存有這種疾病。但是卻有足足1/3的家長拒絕這項檢測,理由不是說他們十分確定自己的孩子絕對沒有X染色體脆弱症,就是擔心這個檢測會對孩子造成太大的壓力。海格曼因這個計劃飽受批評,反對人士群起而攻之。這些人堅信,試圖以駕馭DNA來解決社會問題,無論如何,都會造成「基因極權政治」的危險。
但是,DNA分析使我們的未來也能有更多用比較溫和的方法對抗乳癌的機會,如同密歇根的研究計劃揭露的另一個故事。安妮的一位堂姐得知自己十之八九帶有蹂躪其家族的BRCA1突變,由於她已經有多年沒有照乳|房X光片(諷刺的是,在高危家族裡,這種由於恐懼而造成的疏忽並不罕見),於是她恐慌了。那天稍晚,韋伯安排她照乳|房X光片,結果發現一個初期階段的小腫瘤,它可以輕易切除,但是在例行檢查中肯定會被忽略。自我檢查和定期照乳|房X光片無疑拯救了許多人的性命,但是就一些案例而言,推廣這些檢查的運動有可能造成出乎意料的後果,讓這些人誤以為自己很安全。對有遺傳疾病風險的人進行篩檢,讓我們得以發現誰是那些在做影像檢查時特別嚴格的人。風險愈高,就需要愈仔細的監測。長此以往,我們才可望在愈來愈少的海水中,找到愈來愈多的針。
1999年,蓋辛格聽說賓州大學人類基因療法研究所所長威爾遜(James Wilson)正在進行一個基因療法實驗。蓋辛格罹患鳥氨酸氨甲酰轉移酶(ornithine transcarbamylase, OTC)缺乏症,這種遺傳疾病會破壞肝臟處理尿素的能力,而尿素是蛋白質代謝作用的自然產物。如果不接受治療,這種病有可能致命。雖然它跟苯丙酮酸尿症一樣,可以藉由簡單的藥物治療和適當的飲食來控制,但是OTC缺乏症的確會使病人特別容易罹患其他的疾病。18歲的蓋辛格只是輕微病例,但是幼年一次險些因這病症而死亡的經驗,讓他壯著膽子自願參与進臨床實驗,希望能為自己和其他的病患找到解藥。賓州大學的基因療法目標在於把腺病毒(adenovirus,引起普通型感冒的病毒之一)當做已矯正基因的載體,但是在把攜帶正常OTC基因的病毒注入蓋辛格的肝臟數小時后,他開始發燒,接著發生嚴重的感染,伴隨有血栓和肝臟出血。在注射三天後,蓋辛格不幸死亡。
這個故事始於1934年的挪威。一位年輕的母親決心要找出她4歲和7歲的兩個孩子究竟出了什麼問題,他們出生時似乎十分正常,但是較大的孩子到了7歲還無法完全靠自己上廁所,會說的字也沒幾個,更不用說完整的句子。這個病例引起生化學家及醫師佛林(Asbjφrn Fφlling)的注意。在進行一連串檢驗后,他發現一個他認為跟他們的情況有關的生化異常現象:他們的尿液中含有過多的苯丙氨酸。但他也發現他們並非孤立的個案,他在挪威各地22個家庭里找到的34個相同病例,使他察覺到自己在無意間發現了一種遺傳疾病。
幸好在1972年,新的聯邦政府施行方針重新設計了鐮形細胞貧血症的篩檢計劃,讓篩檢能有效執行,但不會像第一次實施時造成人心惶惶。但是遺傳疾病支持團體失去的信心仍很難彌補;這次經驗讓他們對篩檢計劃始終存有戒心,而對烙印的恐懼也揮之不去。可嘆的是,有時這卻損害到公共健康。
你的DNA可以告訴別人很多有關你的事。如同先前所述,如果你的家族有亨廷頓氏症的病史,你的DNA可以預示你的未來;不久后,DNA或許也可以指出你罹患心臟病等某種常見致命疾病的相對風險,視你是否擁有某個(或數種)特殊的變異基因而定。你擁有哪一型的APOE基因,已經可以作為阿爾茨海默氏症的指標。但是我們該不該擔心,這個極度隱私的信息有一天可能會成為對你不利的工具?對許多美國人來說,他們最擔心的無疑是有一天遺傳信息會讓他們無法參加健康保險。
在英國,30%的唐氏症胎兒是在對5%最年長的孕婦做例行檢查時發現的。從平均每英鎊支出的檢測成果來看,這個方法顯然很有效率(自從當時的首相撒切爾夫人抨擊保健開支后,英國國家保健局經常採取這種計演算法),但其餘70%的唐氏症病例呢?年輕媽媽的胎兒的確比較少見唐氏症,但是這些婦女卻占孕婦總人數的絕大部分。由於以統計上的幾率而言,不值得冒險做這些標準檢查,所以醫界一直想尋找非侵入性的替代指標。最後發現母親血液中有些可偵測的物質能夠提供有用的信息。低α-胎兒蛋白(alpha-fetoprotein, AFP,即甲型胎兒蛋白)和高絨毛膜性腺激素(chorionic gonadotropin)與唐氏症之間有顯著的相關性,但它們並不是三染色體的絕對指標。因此現代的做法是讓比較年輕的婦女做血液篩檢,如果顯示胎兒有唐氏症的可能性,醫生就會建議她們做羊膜穿刺術或絨毛膜絨毛採樣法,以便進行確切的診斷。
阿爾茨海默氏症一般在60歲左右開始發病,但有一種比較罕見,占所有病例5%左右的類型,會在40歲左右就發病。這種早髮型阿爾茨海默氏症會讓病患家屬彷彿置身地獄,就跟亨廷頓氏症患者的親友一樣。早髮型阿爾茨海默氏症患者在生命顛峰時期發病,然後逐漸遭到無情的摧毀。在數代當中有多位成員患病的家族,以前會被描述為遭到毀家滅門的「生物大屠殺」(biological Holocaust)。瑪麗-克萊爾·金在她突破性的乳癌研究中,首度提出一個論點,認為任何疾病的早發形式可能比一般形式具有更清楚的遺傳原因;早期大多數的阿爾茨海默氏症研究就是根據這個論點,把重心放在了早髮型阿爾茨海默氏症上。到了1995年,研究人員已經發現三個相關的基因,它們都跟澱粉樣蛋白(amyloid protein)沉積的處理過程有關,早在1906年,阿爾茨海默醫生(Alois Alzheimer)首度描述這種疾病時,就已經提到病患腦部有澱粉樣蛋白堆積的情形。因此早髮型阿爾茨海默氏症顯然是遺傳的,但是其他比較常見的阿爾茨海默氏症類型呢?
SCID在流行文化中的形象頗耐人尋味。20世紀70年代,這種怪病被拍成了一部賺人熱淚的電視影影片《住在塑料泡泡中的男孩》(The Boy in the Plastic Bubble)。到了90年代,泡泡男孩(Bubble Boy)竟成為情境喜劇《歡樂單身派對》(Seinfeld)中的搞笑角色。2001年,迪斯尼發行了一部庸俗的電影,內容是一個被困在泡泡里的男孩一連串愚蠢的冒險行動,片中沒有指明他得的是哪種病,但明眼人一看就知道。科學在面對這類可怕的疾病時根深蒂固的無力九*九*藏*書感,多少解釋了為何原先的苦情劇會演變成苦中作樂的喜劇。但是對患者及其家屬來說,這種無力感只是令人更加難受。特別是會造成身體日益衰退且無法挽回的疾病,只要一被診斷出來,無異於宣判死刑。在療法付諸闕如的情況下,有些人寧可不知道自己可怕的命運,特別是在看到摯爰的親人遭受病魔的摧殘后。以上一章看到的南西·魏克斯勒為例,她得亨廷頓氏症的幾率是50%,而且這種病已經奪走她的母親和舅舅。南西為了找出這種遺傳疾病的元兇,在馬拉開波湖和美國的遺傳學實驗室經年累月地辛勤工作。然而即便她卓絕的奮鬥最後終於得以分離出這種病的基因,找出致命的突變,但是找到治療法的希望依舊渺茫。儘管她已經盡了最大的努力,讓診斷用的基因檢測法得以問世,她本人仍然表示不會接受檢測,至少在可行的療法有一線曙光之前不會。她寧可活在不確定中,也不願知道這場輸贏幾率各半的賭局結果:她有50%的幾率得面對身心衰退的噩運,這位活力充沛的女鬥士,有朝一日可能只剩下一具毫無生氣的軀殼。
至今「正義世代」計劃已經篩檢超過7萬人,發現超過100對有風險的情侶。這個計劃使泰賽二氏病例不斷減少,看來它是大大成功,但是仍有一些猶太人譴責這個計劃。他們認為這個計劃呼籲所有的年輕人接受篩檢是一種壓迫手段,同時認為它極力建議某些人重新考慮結婚決定的做法,是一種威嚇。反對者把艾克斯坦的改革做法說成是「優生學」(對猶太族群而言,這是最讓他們痛苦的一個字眼),但是這種具有煽動意味的說法根本無法動搖一個重要的事實:這個計劃明顯在它服務的小區獲得熱烈的支持,因為小區成員很清楚泰賽二氏症的可怕。事實上,艾克斯坦證明了篩檢計劃可以有效地進行,同時兼顧傳統文化,甚至在社會習俗及宗教戒律與遺傳篩檢的原則看來難以兼容的情況下,也能行得通。
許多女性讀到這裏可能會產生一個問題:我懷孕時為什麼沒有做纖維囊泡症、X染色體脆弱症或假肥大型肌營養不良症的篩檢?遺憾的是,這些婦女中有的人可能已有孩子罹患這類疾病。在使醫學技術大轉型的遺傳學革命之後,有一個格外令人沮喪又沒有道理的事實:科學進展與病人醫護的脫節。事實上,比較正確的說法應該是,這點從未得到應有的重視,讓兩者立即掛上鉤。無論如何,許多婦女根本就沒有機會得知她們有哪些選擇,而現在已經可用的檢測方法,使用率也極低。
但是,儘管有這些技術上的困難,纖維囊泡症的產前篩檢仍可以找出許多已經有這種病的胎兒。既然如此,為什麼沒有擴大實施篩檢?矛盾的是,造成篩檢範圍僅限於有纖維囊泡症病史家族的主要推手,正是支持纖維囊泡症患者的團體。他們擔心擴大篩檢範圍會瓜分有限的資源,使尋找療法的終極目標獲得的資源變得更少。這樣的擔憂是可以理解的,特別是現在。據估計,目前約有3萬名美國人是纖維囊泡症患者。醫療上的進展已大幅延長了患者的預期壽命,而找到療法應該也是可以期待之事。儘管如此,我們還是不能說療法即將問世,這是不負責任的說法。患有纖維嚢泡症的嬰兒,仍可能得和這種使身體日益耗弱的疾病奮鬥一生。儘管治療纖維囊泡症絕對是首要之務,但仍應讓有意願的孕婦有接受篩檢的途徑。然後她可以在對胎兒狀態有充分了解的情況下,自由作出適當的選擇。
但是我們仍舊有個問題必須解決,亦即如何鎖定那些受突變影響的細胞,也就是需要換裝「替代基因」的細胞。如今這仍是基因療法所面臨的最艱巨挑戰。如何把好基因植入肌肉細胞,以便治療假肥大型肌營養不良症?如何把好基因植入肺細胞和腦細胞,以便治療纖維囊泡症和亨廷頓氏症?因此,在首度進行基因療法的實驗時,以鮮為人知的腺苷脫氨酶缺乏症為目標,是很合理的選擇。因為要治療這個病症所需鎖定的目標細胞,是在血液中循環的免疫系統細胞,很容易取得。安德森的研究小組從這兩個女孩的血液中取得大量的免疫細胞,在培養皿中培養,再讓它們接觸攜帶了正常基因的反轉錄病毒。等這些細胞原有的DNA和攜帶替代基因的病毒基因組結合后,再把這些細胞重新注入病人的血液中。
阿善蒂和辛迪的實驗並不是美國國家衛生研究院第一次插手基因療法領域的案例。事實上,該院的人類基因療法小組委員會正是為了回應第一個基因療法實驗,而在1980年成立的。那次實驗不僅失敗,還引起爭議,以至於美國政府幾乎要扼殺掉這個才剛剛起步的領域。
有8個細胞的胚胎
在我們的文化中,人類的生殖生物學似乎是無窮爭議的來源,而跟操控人類胚胎有關的做法,無論目的為何,勢必成為攻擊的焦點。胚胎著床前遺傳診斷術也不例外,然而,撇開道德考慮不談,這個程序仍有兩大缺點:接受診斷術的夫婦必須有堅定的決心;此外,就跟所有形式的試管受精—樣,這種技術非常昂貴。但基本上它的成效很好,造成的傷害也比墮胎少得多,現在只希望日後能有更好的技術問世,使成本跟著降低,這是科技發展的常見模式。在我們對抗遺傳疾病的戰爭中,胚胎著床前遺傳診斷術可能將是極重要的武器。
移植療法的進步看似充滿希望,1983年10月,就在戴維過了12歲生日的一個月後,他接受姐姐捐贈的骨髓,動了骨髓移植手術。不幸的是,後來發現姐姐的骨髓內含有一種病毒,在戴維毫無防衛能力的系統里引起了惡性淋巴瘤。1984年2月,他被迫離開「泡泡」,住進加護病房。他在不久後去世,但至少在最後一段日子里,他終於能夠體會到人類溫暖的撫觸。
從遺傳學的觀點來看,目前為止我們討論過的疾病都還算「簡單」:它們都是由單一基因的突變所引起的,環境對一個人是否會罹患這些疾病毫無影響。但是癌症這類疾病則複雜得多,如同前述,癌症有可能在遺傳與環境的合併影響下發生。但是即使是癌症,也有影響特別大的基因存在,無論環境的影響為何。雖然與乳癌有關的基因之一BRCA1,只佔所有乳癌病例的5%,但是根據估計,這個基因發生突變的婦女到了60歲時,會有90%的幾率得乳癌。
第九章提過的德系猶太人,也很清楚致命的突變會對在遺傳上與他族隔離的族群造成什麼後果。泰賽二氏症這種可怕的疾病在德系猶太族群中的發生率,比大多數的非猶太族群高100倍。罹患泰賽二氏病的嬰兒在出生時看似正常,但後來他們的發育會逐漸變慢,日漸失明。到了2歲左右,他們會發作癲癇,情況持續惡化直到死亡,通常是在4歲之前,而且死亡時大多目盲並癱瘓。血紅素疾病在特定族群中相對比較常見,通常可以用這是為了對抗瘧疾而產生的適應保護來解釋,但是泰賽二氏病不同,這種病在德系猶太族群中的高發病率,至今依舊是謎。遺傳瓶頸或許是罪魁禍首:這種突變可能起先就存在於創始族群里相對較少的一群人身上,這群人在猶太人近代遠離故土的第二次「大流散」期間分支出去,成為現今的德系猶太人。類似的現象或許也可以解釋為何這種突變在魁北克西南部的法裔加拿大人,以及美國路易斯安那州的法裔卡真人(Cajun)當中格外常見:在他們所源出的一小群創始族群中,剛好有這種不幸的突變存在。另一個解釋是:這個隱性基因(只有一份泰賽二氏症突變)的帶因者對肺結核的抵抗力可能較強,這對歷來傾向於以人口密集的都會區為家的歐洲猶太人來說,或許是一個優勢。
支持纖維囊泡症患者的人一向盡量防止患者被打上某種歧視烙印,他們擔心篩檢會間接導致這個結果。事實上,在遺傳篩檢史上的確有過不幸的前例,令所有的病患支持團體難以忘懷。早在DNA篩檢時代來臨之前,最早期針對遺傳疾病所作的診斷之一,是為了檢測鐮形細胞貧血症而發展出來的,在美國,罹患這種疾病的主要族群是非洲裔的美國人。如同第三章所見,那些同時擁有兩個突變的「鐮刀狀」血紅素基因的人,得承受身體逐漸耗弱的痛苦,而只有一個這種基因的人(亦即帶因者)則幾乎不會受到任何影響。
羅塞斯是神經學家,對假肥大型肌營養不良症等肌肉疾病學有專長,他在1984年開始研究晚髮型阿爾茨海默氏症。1990年,他宣稱第19號染色體上的一個基因顯然與這種病有關,但這個說法遭到質疑。最令羅塞斯高興的事,莫過於證明別人都錯了。兩年後,他真的找出了這個重要的基因。最後發現它編碼載脂蛋白E (apolipoprotein E, APOE),這種蛋白質與膽固醇的處理過程有關。這個基因有三種形式(即有三種等位基因),APOEε2、APOEε3和APOEε4,但是後來證明具有決定性角色的是APOEε4:只要有一份這個變異,一個人罹患阿爾茨海默氏症的幾率就會增加四倍。若是有兩份,則患病幾率會比沒有APOEε4等位基因的人高10倍。羅塞斯發現有兩個APOEε4的人當中,55%會在80歲前發病。這種相互關係是否可以作為進行遺傳檢測的基礎?可能不行。儘管與這個疾病有關,APOEε4等位基因仍很常見,要作為檢測阿爾茨海默氏症的指標,它還不夠準確。雖然有APOEε4等位基因的人罹患阿爾茨海默氏症的可能性較高,但還是有很多有兩個APOEε4等位基因的人沒有發病。但是運用APOEε4篩檢,再配合臨床評估,的確提高了診斷阿爾茨海默氏症的準確度。或許等我們了解了它們之間的因果關係后,就可以改良遺傳分析技術。最近在實驗鼠身上引發阿爾茨海默氏症的研究顯示,APOE與造成阿爾茨海默氏症患者神經細胞死亡的蛋白質的代謝作用有關。
如同先前所見,儘管X染色體脆弱症是性聯遺傳,但男性和女性都有可能患病。因此,這種病自然會成為以特定基因為主的胚胎著床前遺傳診斷術的目標,但是父母的態度仍要非常積極,而且明白扶養X染色體脆弱症的兒童非常辛苦,才能說服醫生執行這個診斷術。
唐氏症的發生率會隨母親年齡的增加而升高。20歲的母親產下唐氏症寶寶的幾率大約是1/1700,但是35歲的母親產下唐氏症寶寶的幾率升至1/400,45歲的母親則升至1/30。基於這個原因,許多高齡產婦選擇對胎兒作產前診斷,判定其第21號染色體是否有三條。這個檢測首次執行是在1968年,今日它已成為超過35歲的孕婦需接受的例行檢查。
過去10年來,很少有疾病像阿爾茨海默氏症一樣,讓那麼多人感到恐懼,每年因這種病而身心遭受折磨的人愈來愈多,現在有超過400萬名美國人罹患這種病。患者的親友首先會注意到他們犯了輕微的失憶,像是想不起來最近的事情或找不到正確的字眼,而他們可能會把這些情形歸咎於年紀大了。接著患者的情緒可能有明顯變化的跡象,但這在老年人身上也並不算不正常。但是隨著病程發展,病人的癥狀會日益明顯,讓人不至於再弄錯。喪失記憶的情形很快會變得異常嚴重,造成他們無法處理平時熟悉的工作,甚至簡單的家事。語言愈來愈不流暢,經常說到一半就因為思緒中斷而停下來。患者在知道自己的這些改變后可能會罹患憂鬱症,使日益惡化的性格改變問題更加嚴重。重度阿爾茨海默氏症患者不知道自己是誰或身在何處,甚至認不出最親近的人。隨著記憶與性情不斷惡化,他們所擁有的個人特質也逐漸被摧毀。
遺傳知識會造成道德上的兩難困境。凱瑟琳先前並未被告知羊膜穿刺術有可能查出三染色體21以外的情況,或許細胞遺傳學家逾越了職責範圍,他們應該僅針對當初做這個篩檢的目的提出報告。當然,如果臨床醫生使用的是HSH法,就不會有這種問題,畢竟FISH法只能查出第21號染色體的數目。隨著基因篩檢法日益精進,就像打開潘多拉的寶盒一樣,篩檢結果的影響會遠超過當初做這個篩檢的目的,有時影響所及甚至不僅那些做篩檢的人而已。在這方面,最明顯的實例莫過於對有遺傳病史的家族所做的基因篩檢,例如有假肥大型肌營養不良症或纖維囊泡症的家族。在這些例子中,診斷不是由細胞遺傳學家來做,而是分子生物學家,他們分析的不是一段段染色體,而是特定區段的DNA。他們先用羊膜穿刺術從胎兒身上取得組織樣本,兒童或成人則從抽血或用壓舌板從口腔內部颳得的細胞,取得組織樣本,再從這些樣本中萃取出DNA。現在這些檢測通常會用PCR法擴增DNA樣本的重要區段(可能有問題的基因read•99csw.com),然後再作一系列的分析,以判定這個基因是否有突變。針對任何一人所做的檢測結果,都可以告訴我們一些有關其親屬的基因狀況。
倫敦漢默史密斯醫院(Hammer smith Hospital)的溫斯頓(Robert Winston)是一流的婦科顯微手術醫生,擅長於輸卵管缺陷矯正手術(輸卵管缺陷會使婦女無法受孕)。他也是在英國電視上推廣科學與生物醫學研究的健將,甚至有餘暇以勛爵的身份擔任上議院議員,就相關議題提供意見給政府。他結合試管受精(IVF)與以PCR為主的DNA診斷兩種尖端技術,發展出一種方法,在胚胎殖入婦女的子宮並開始發育前,就檢查這個胚胎的基因狀態。數個胎體在試管里受精后,在實驗室里發育,直到每個受精卵分裂三四次,有8到16個細胞為止。這時小心地從每個胚胎移出一兩個細胞,取出DNA,再用PCR擴增相關的序列,以決定每個胚胎是否存有突變。PCR能擴增極微量的目標DNA,多虧了它這驚人的能力,這種能在胎兒發育極早期就進行診斷的方法才得以問世。然後父母便可以放心地殖入沒有遺傳疾病的胚眙。
在主持人類基因組計劃時,我必定會提撥經費,協助民眾了解定序機器即將透露的知識會對眾多人的生活造成什麼影響,無論這些影響是好是壞。在提撥總預算的3%(後來是5%)用於這方面后,我任命亨廷頓氏症專家南西·魏克斯勒主持一個稱為ELSI的小組委員會,專責探討我們的研究在道德、法律及社會層面的影響。ELSI的主要計劃之一是一系列有關遺傳篩檢的試驗性研究。在每個新生兒都要接受苯丙酮酸尿症篩檢的時代,我們的確有必要質疑:在面對纖維囊泡症、假肥大型肌營養不良症、X染色體脆弱症和其他科學可以預測的嚴重疾病時,醫學連篩檢這些疾病的選擇都無法提供,是否是負責任的做法?當時是90年代早期,而今我們仍然停留在試驗性階段,除了一些零星的小規模研究,幾乎毫無進展。造成這種停滯狀態的原因有現實的鈔票問題,也有人類生命的本質與尊嚴等深奧的哲學歧見。簡而言之,這當中牽涉所有伴隨遺傳學革命而生的社會現象,從謀取經費到對生命的集體省思都有。目前有關假肥大型肌營養不良症和亨廷頓氏症的篩檢,一般僅用於已經有一名成員發病的家族。這個限制的根據在於這些疾病很罕見,而這類篩檢的費用很昂貴。這種社會成本計演算法是有爭論的餘地,但相同的論據並不適用於纖維囊泡症的情況,儘管如此,纖維囊泡症的篩檢仍有限制。先前提過,纖維囊泡症的發生率是1/2500,這使它成為最常見的遺傳疾病之一,在北歐人之間尤為常見。有鑑於這種疾病的遺傳缺陷是發生在第7號染色體,而且是必須同時有兩份突變才會發病的隱性遺傳模式,就令人覺得這種病的發生率實在高得驚人。只有一份突變的人不會發病,但會成為帶因者,有可能把突變傳給子女。根據流行病學的調查和估算,每25位歐洲血統的美國人當中,就有一位是帶因者。
在這類遺傳疾病中,至今最廣為人知的是以英國醫師唐恩(John Langdon Down)之名命名的唐氏症候群(Down syndrome)。1866年,唐恩在擔任智能障礙療養院的醫務總監時,首度描述了唐氏症的特徵。他注意到在他的療養院里,10%的病人長得很像:「這情形實在太明顯,在讓他們並排站立時,很難相信他們不是手足。」然而一直到20世紀90年代后,法國醫生勒熱納(Jérome Lejeune)才深入研究了這種疾病的生物學原理,他發現唐氏症兒童的某染色體有三條,後來發現是發生在第21號染色體。正常的情況是有成對的兩條染色體,稱為雙染色體(disomy),所以唐氏症候群在遺傳學上的說法是「三染色體21」(trisomy21)。
得知胎兒有唐氏症的婦女通常都會選擇終止懷孕。因此,在實施例行產前篩檢的國家,唐氏症寶寶出生的數目日漸降低。然而在統計上,實情比這複雜:婦女晚生子女的趨勢(經常是基於工作原因)其實增加了婦女懷唐氏症胎兒的幾率。所以,在英國是以該年所有孕婦的年齡來預估可能會有的唐氏症寶寶數目,再以此為基準來衡量篩檢計劃的成效。現在我們看到的是唐氏症寶寶的比例不斷下降;例如在1994年,篩檢計劃使唐氏症寶寶的發生率減少了40%左右。
20世紀50年代,用顯微鏡來研究染色體的細胞遺傳學(cytogenetics)興起。把這方法用於診斷後,科學家很快就發現,染色體數目異常(通常是多一條或少一條)必定會引起嚴重的機能障礙。這些問題根源於基因數不平衡,正常狀況應該是每個基因有兩份。這類情況不像假肥大型肌營養不良症或纖維囊泡症會在家族裡遺傳,但仍舊屬於遺傳的範疇:它們是因為產生精|子與卵子的細胞分裂過程發生意外,而自發形成的疾病。
以針對亨廷頓氏症所做的檢測為例,在最近一個例子中,有一位20多歲的男士到提供遺傳篩檢(基因篩檢)的診所,要求做亨廷頓氏症的檢測。他的祖父是因這個疾病過世,而他40多歲的父親跟南西·魏克斯勒一樣,選擇生活在50%得病率的陰影下,不願知道確切的結果。由於亨廷頓氏症是在人生中較晚的時期發病,因此很有可能這位父親已帶有突變,只不過還沒有顯現任何癥狀。這位年輕人知道自己帶有這種突變,並在未來發病的幾率是1/4,但他想知道確切的結果。問題是,如果他發現自己的確有這種突變,那麼他必定是從父親身上得到的,這代表他父親未來一定會發病。這個兒子要求獲知遺傳信息的想法,跟他父親不想得知真相的意願發生衝突,家庭間起了紛爭。最後是在這位年輕人的母親介入下,他才沒有進行篩檢。她的論點是,她丈夫有權不想得知類似被判死刑的真相,相較之下,她兒子想得知真相的意願自然較不重要。這個極端的例子凸顯了基因診斷與其他診斷的不同之處。我從自己的基因得知的事,有可能對我的血親造成影響,無論他們本身是不是想知道。
只要我們一直停留在目前的「中間階段」,亦即大致上只有診斷能力而沒有治療能力,那麼基因知識仍會顯得可怕。但這並不是我們第一次遇到這種醫學困境。在20世紀初也有類似的情況,當時被診斷出糖尿病的嬰兒等於被宣判死刑。如今隨著胰島素療法的問世,這類兒童可以好好地長大成人。我們作研究的目的就在於希望不久的將來,對於亨廷頓氏症這類疾病的診斷能有相同的結局:從宣判死刑變成開立療方。
治療方面呢?我們在治療大多數的遺傳疾病上,進展仍跟亨廷頓氏症一樣令人沮喪。我們已經有足夠的知識可以進行診斷,或許也能避免它們發生,但還沒有找到治療方法。幸好遺傳學知識已讓我們找到一些疾病的療法,只可惜這些療法中極少像苯丙酮酸尿症的治療法一樣簡單有效——苯丙酮酸尿症患者可以通過限制一些飲食項目,恢復正常的生活。
遺傳疾病經常造成特定組織的細胞逐一死亡,如假肥大型肌營養不良症的肌肉細胞、亨廷頓氏症和阿爾茨海默氏症的神經細胞。對於這種在不知不覺中惡化的疾病,目前仍沒有權宜之法。雖然幹細胞療法目前仍在早期發展階段,但我相信最終我們利用幹細胞來治療這類疾病的機會是很高的。人體內大多數的細胞只能自我複製,例如肝細胞只能製造肝細胞,但是幹細胞可以製造多種類型的特化細胞。在最簡單的例子中,一個剛受精的卵子(潛力最大的幹細胞)最終可以製造出所有216種已知的人類細胞。因此幹細胞最容易從胚胎中取得;它們也存在於成人體內,但成人的幹細胞不像胚胎的幹細胞能分化成任一類型的細胞。我們才剛開始了解如何誘導幹細胞製造特定類型的細胞,希望未來我們可以用健康的新細胞替換亨廷頓氏症和阿爾茨海默氏症患者失去的腦細胞。但是我要提醒,我們仍須經過漫長的努力,才能徹底了解使一個細胞朝特定方向發展的分子啟動機制。我們大約還要10年左右時間來解決這個發育生物學上的問題,才有辦法妥善研究幹細胞的治療價值。如果這類研究因宗教考慮而受阻,這對科學及所有可能受益於幹細胞療法的人來說,都是一個悲劇。民意調查一貫顯示,大多數美國人支持使用胚胎的幹細胞來進行研究,然而政治人物卻持續迎合少數大聲反對的宗教團體,結果在美國形成立法加以限制,阻礙了這類具有極大潛在價值的技術發展。
事情也如預期發展:10個月後,兩名病童身上都能找到含有一份正常基因的T細胞,而他們的免疫系統功能也跟正常兒童一樣。費希爾的療法後來也被用於其他的SCID病童。在經過漫長而且一波三折的開端后,基因療法終於獲得明確的成功。但是這場香檳慶祝會的氣氛沒有持續多久。2002年10月,醫生髮現最早接受這種治療的兩個病童中,有一個患了白血病,這種骨髓方面的癌症會使特定類型的細胞生產過剩。儘管目前為止無法確認這是否是基因療法所造成的,但是間接證據仍然很有說服力。基因療法似乎治好了這名嬰兒的SCID,卻引起白血病的副作用。
幸好SCID很罕見,但是兒童發生遺傳疾病的幾率卻十分驚人。事實上,大約2%的新生兒天生有嚴重的遺傳異常。根據估計,在兒童醫院的住院病例中,1/10與基因有直接關係,而半數左右與基因也有間接關係。遺憾的是,戴維的例子顯示出我們現有的知識對大多數遺傳疾病力有未逮之處:我們知道哪裡出了問題,也能診斷,但是在治療方面,我們能做的相對很少,遑論治愈。
在倫敦的希臘塞普勒斯裔族群中,地中海型貧血的帶因者高達17%,相當驚人。嚴重的地中海型貧血是傷害力最大的血紅素疾病,會造成畸形的、有時甚至會結核的紅血球,引起肝臟和脾臟腫大,患者通常無法活到成年。1974年,皇家自由醫學院(Royal Free Medical School)的摩戴爾(Bernadette Modell)展開一個有系統的篩檢計劃,結果這個計劃受到倫敦塞普勒斯族群的熱烈歡迎,因為他們太清楚這種病的嚴重性,長久以來他們的小區一直深受其害。義大利薩丁島(Sardinia)1974年也開始進行類似的計劃,使血紅素疾病的發生率從1/250大幅減少到1/4000。
對於證實胎兒患有遺傳疾病的孕婦而言,產前篩檢提供了明確的選擇:墮胎或繼續懷孕。羊膜穿刺術只能用於至少已經15周大的胎兒,這個限制讓選擇墮胎更為痛苦。在這個階段,墮胎所抹殺掉的不是一團沒有特徵的細胞,而是一個小生命——在超音波影像的協助下,胎兒顯得非常真實,足以讓父母對這個發育中的胎兒產生親情。如果基於遺傳篩檢的結果而必須作出選擇的話,大多數的父母——至少是那些並非堅決反對墮胎的人——絕對寧可在胎兒發育更早期的階段作選擇。這就是發明胚胎著床前遺傳診斷術(preimplantation diagnosis)的靈感來源。
血紅素疾病(hemoglobinopathy)是由血紅素分子的機能障礙所引起的疾病,包括多種地中海型貧血(thalassemias)和鐮形細胞貧血症在內,一般認為它們是最常見的遺傳疾病,全球人口大約有4.5%帶有其中一種疾病的突變。先前已經談到,鐮形細胞基因具有抗瘧疾的特質,所以在瘧疾肆虐的地區會受到自然選擇的青睞。因此,這種突變的高出現率最初僅見於世界上的這類地區。其他類似的血紅素疾病分佈模式,也是相同的適應優勢所造成的。醫學界早就知道,某些突變在一些民族中比較常見,無論屬於這個民族的人身在何處。
新基因要怎麼注入病患體內?在當時,反轉錄病毒(retrovirus)似乎是合理的選擇。一般來說,病毒是有效的基因載體,它們靠把自己的DNA注入其他的細胞來生存。反轉錄病毒是一群特殊的病毒,擁有的九九藏書遺傳物質是RNA,不是DNA。大多數的病毒在感染一個細胞后開始繁殖,然後殺死宿主細胞,「子代」的病毒逸出后再去感染其他的細胞。但是反轉錄病毒一般對宿主細胞比較仁慈和溫和,新的病毒複本會自然離開,並不會摧毀宿主細胞。這並不是說反轉錄病毒對宿主生物體的傷害較小;有時恰恰相反,艾滋病毒HIV就是一個實例(HIV或許是最廣為人知的反轉錄病毒)。但這的確代表病毒基因(以及這個病毒可能載入的額外基因)會永久成為未被摧毀的細胞基因組的一部分。遺傳工程已能製造出足夠安全的反轉錄病毒,供基因治療使用;在除去病毒中所有將被入侵的宿主細胞基因組所不需要的基因后,反轉錄病毒就成為了理想的基因載體。
1968年,在發現泰賽二氏症患者的紅血球細胞中有過多的神經節糖RGM2(ganglioside GM2)后,泰賽二氏症的原因終於大白。神經節糖RGM2這種化學物質是細胞膜的基本成分,在正常人體內,多餘的神經節糖RGM2會被一種重要的酶分解為相關的化合物,但是泰賽二氏症患者缺乏這種酶。1985年,梅洛威茲(Rachel Myerowitz)和她在美國國家衛生研究院的同事分離出了為這個酶編碼的基因,證明泰賽二氏症患者的這個基因的確發生了突變。
通過這種生化途徑來矯正基因異常情況的做法,顯然可行又有效。但是即使重組法的效率驚人,這種治療法仍很昂貴,每年要17.5萬美元,而且必須持續補充蛋白質,對病人來說也是一種負擔。在這種情況下、遺傳學家自然會希望能找到一種實際可行的方法來除去病根,而不是僅反制突變所造成的作用而已。遺傳疾病的理想療法是改變遺傳,亦即矯正造成問題的基因。這種基因療法可以讓病患終生受益,一旦矯正好基因,就可以持續一生。至少在原則上有兩種方法:體細胞基因療法(somatic gene therapy),亦即改變病人體細胞的基因;生殖細胞療法(germ-line therapy),亦即改變病患精|子或卵子內的基因,防止有害的突變傳給下一代。
看來基因療法似乎還要很長的時間,才能創造基因革命開始時預見的奇迹。蓋辛格死亡事件是一次嚴重的失敗,但是SCID療法的白血病副作用造成的傷害甚至更大。在蓋辛格的個案中,主因在於不可原諒的管理疏失,這個問題通過更嚴格的規定應該已經解決了。但是副作用的問題至今還沒有可行的解決方法。在這個例子中,或許我們只能無奈地自我安慰:至少基因療法治好了SCID,起碼SCID是比這療法引起的白血症還嚴重的病。幸好那名罹患白血病的男嬰對化學療法的反應顯然良好。然而,蓋辛格和白血病的事件具體呈現出了許多困難的問題,如果體細胞基因療法要成為醫學主流,就必須先解決這些問題。而我也不會天真地以為,未來的實驗就不會遇到更多的困難。或許還要一段時間,我們才能確切地宣稱已經解決掉所有想像得到的危險。儘管如此,我仍舊認為,這個技術破解遺傳疾病宿命的潛力實在很大,醫學界絕不能放棄它。
從那時起,我們就可以根據這個基因,對明確的目標族群進行十拿九穩的產前檢查,而這也正是最適合實施篩檢計劃的情況。但是在遇到陽性的診斷結果時,產前篩檢惟一能提供的補救方法就是墮胎,而德系猶太族群——至少是那些嚴守教規的正統派——是禁止墮胎的。幸好,篩檢也可以找出打算結婚生子的人是否身為帶因者,所以對虔誠的教徒來說,符合道德觀念的可行方法是以準備成婚的男女為篩檢對象。紐約猶太教教士艾克斯坦(Yosef Eckstein)的10個孩子中,有4個死於泰賽二氏症。1985年,他設立了「正義世代」(Dor Yeshorim)計劃,在當地的正統猶太教小區推動泰賽二氏症的篩檢。他們鼓勵年輕人在高中和大學時接受免費檢測,這個計劃的特殊之處在於高度保密:就連那些接受測試的人也不會得知自己是否是帶因者,而是只拿到一個密碼。未來等兩個人打算結婚時,他們可以各自打電話給「正義世代」並提出密碼。惟有在兩人都是帶因者的情況下,「正義世代」才會透露狀況,並且提供諮詢。這種有必要才告知的保密方式,目的在於避免讓帶因者被打上烙印,同時仍能對抗泰賽二氏症的威脅。

6歲的唐氏症兒童JD和他的父親
就目前言,在遺傳疾病的治療上,我們還沒有辦法以幹細胞療法來更替大量的細胞,但是或許可以補充一種失去的蛋白質。發生率為四萬分之一的高雪氏症(Gaucher disease)是一種罕見的疾病,起因於葡萄糖腦苷脂酶(glucocerebrosidase)的基因發生突變。葡萄糖腦苷脂酶協助分解一種特殊的脂肪分子,若是沒有它們,這種脂肪分子會在人體細胞里堆積,造成傷害。高雪氏病有可能造成嚴重的傷害,其癥狀包括骨頭疼痛與貧血。早在1974年,醫界就開始嘗試直接補充病人喪失的這種酶。結果證明這種療法很有潛力,但它的準備工作卻是一場惡夢:用來補充的酶必須從人類的胎盤中取得,而從兩萬個胎盤中取得的量,只夠一個病人一年所需。在20世紀90年代初,研究人員有了重大突破,合成出這種酶的改良形式,讓最需要它們的細胞更有效地吸收。1994年,基酶(Genzyme)生技公司開始運用DNA重組法製造這種改良型的酶。高雪氏症的治療法不是解決造成這種病的遺傳根源,而是反制這種突變所造成的效果,把缺陷基因無法提供的重要蛋白質提供給病人。
現在我們已知PKU是編碼苯丙氨酸羥化酶(phenylalanine hydroxylase)的基因發生突變聽引起的,這種酶負責將苯丙氨酸轉變成另一種氨基酸,即酪氨酸(tyrosine)。PKU是一種罕見的疾病,在北美的罹患率大約為1萬人中有1人,而且具有隱性遺傳模式,孩子要從雙親遺傳到兩份突變基因(父一份,母一份)才會發病。病童的羥化酶無法發揮功能,造成苯丙氨酸在血液中累積,傷害腦部發育及導致嚴重的智力障礙。預防方法很簡單:PKU兒童只要從出生起即以苯丙氨酸含量低的飲食為主,就可以正常成長,也就是盡量少攝取蛋白質,不喝含人工糖精的汽水,這些是苯丙氨酸的兩大主要來源。僅是營養來源一項就足以造成腦部正常發育和嚴重殘障的差別。從兒童一出生就檢測其PKU狀況,當然很重要。美國醫師蓋斯瑞(Robert Guthrie)發明了一種簡單的檢測方法,可以得知血液中苯丙氨酸的含量,他積極推廣這種方法,直到它成為新生兒的標準檢測項目為止。自1966年起,每個新生兒都由腳跟抽血採樣,分析苯丙氨酸含量。每年有幾百萬名新生兒接受蓋斯瑞血液檢測法,不需要檢驗DNA任一鹼基對,就可以篩檢出這種遺傳疾病。在這個檢測計劃推行前,美國大約有1/100智障兒是PKU所引起的,現在每年只有少數病例而已。
在克萊恩事件后,仍然有嘗試基因療法的科學家因違反規則而惹上麻煩。遺憾的是,一直要到一位嘗試基因療法的病人死亡,人們才深切地體會到,基因療法這種涉及病毒、生長因子和病人的複雜治療方法是有危險的。此外,由於基因療法有許多未知因素,因此所有與人類有關的程序,絕對有必要接受嚴格的監督。亞利桑那州少年蓋辛格(Jesse Gelsinger)之所以死亡,不僅是因為我們具備的知識尚不足以有充分把握來預測一個人對基因療法的反應,也因為科學家走了不能原諒的快捷方式。
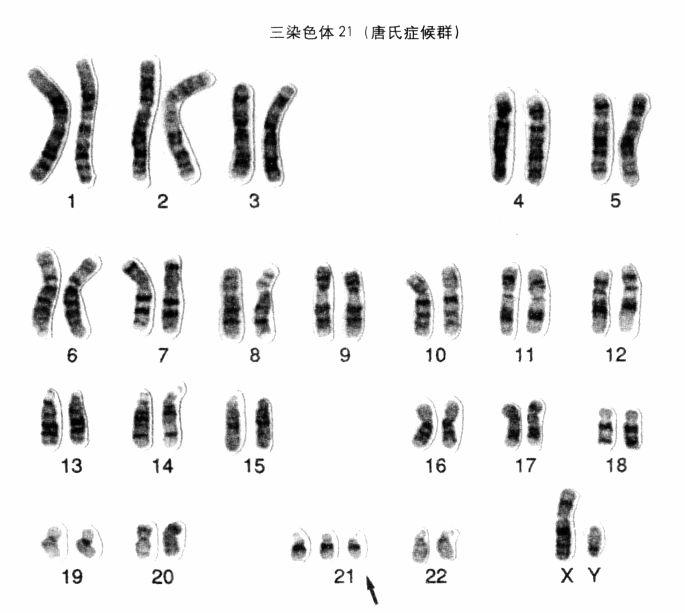
一名男唐氏症患者的染色體組型(karyotype),即全數染色體。從圖上可以看出,他的第21號染色體多出一條,成了「三染色體21」。

戴維·維特因遺傳性免疫系統疾病而極容易發生感染,他從小在隔離的無菌世界「泡泡屋」長大,成為最早的「泡泡男孩」。
同時,或許我們可以欣慰地得知,儘管保險業已經有大量可以自由使用的基因資料,而且無論他們給民意調查的答案是什麼,整體來看,保險公司至今似乎還不會衝動地把遺傳基因列為保險費率的計算因子。我遺傳到的皮膚極白,現在也已證明這樣的膚色容易得癌症,但我上次檢視保單時,並沒發現我的保費因為這個因素而增加。同樣地,這當中的原因並不在於心懷慈善,而在於商業考慮。保險業者一向利用精算表來設定保險費率,這種精算表則是根據我們的生活方式來估算整體的健康狀況與壽命。我想就算基因數據到處都可取得,保險業者還是會發現生活方式的因素,例如一個人抽不抽煙,是在煤礦還是花店工作等等,更能預測一個人的健康風險,遠勝過絕大多數由遺傳變異所決定的細微個人差異。那些從DNA得知自己未來的健康必然會逐漸衰退的人,無疑需要法律的特別保護。但是容易患心臟病和癌症的體質勢必會很普遍也很複雜,因此要以它們作為縮減成本的基礎並不切實際。所謂保險就是以大多數沒有病痛、不必申請理賠的人所付的保費,來救助少數不幸者,這樣的基本前提不太可能因為獲得了更多遺傳信息就廢除掉。

史蒂文森夫婦和子女合影:他們的長子特勒有X染色體脆弱症。胚胎著床前遺傳診斷術確保他們的幼|女莎曼莎沒有這種疾病。
CNBC電視新聞記者黛比·史蒂文森(Debbie Stevenson)的兒子特勒(Taylor)患有X染色體脆弱症,但一直到她的第二個兒子詹姆士(James)出生后才診斷出來。儘管在50%的得病率,詹姆士卻幸運地沒有得病,但史蒂文森家仍不願把第三個孩子的未來交由命運來決定。他們決定藉助胚胎著床前遺傳診斷術,黛比表示:「有些人認為選擇健康的胚胎是不道德的,但我想這總比在得知你的寶寶有重大疾病時,才作是否要墮胎的痛苦決定來得好。」這個家庭在花了一整年的時間尋找到願意執行這個技術的實驗室后,黛比終於在2000年如願通過這個技術懷了史蒂文森家最小的成員。在做過X染色體脆弱症的檢査后,小寶寶薩曼莎(Samantha)證明跟詹姆士一樣,沒有遺傳到特勒的可怕疾病。

目前還是好消息:費希爾(左)和他的合作夥伴卡瓦沙那-卡佛(Marina Cavazzana-Calvo)在2000年4月宣布他們突破性的基因療法獲得了成功。
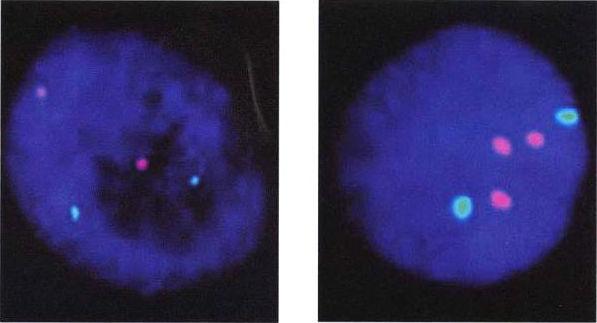
用熒光染色來測定染色體數。一個細胞核(深藍)的第10號(淺藍)和第21號(粉紅)染色體接受檢測。左圖的影像是每對染色體各有兩條的正常染色體組型,右圖是唐氏症染色體組型,其第21號染色體多了一條。
三染色體的情況也會發生在其他的染色體之上,但是這些情況所造成的結果太過嚴重,因此孕婦都會自然流產,只有第13號和第18號染色體的三染色體情況例外。但是有三染色體13的嬰兒大多隻能活數周,而有三染色體18的兒童通常會在1歲前死亡。包括三染色體在內的染色體異常可能經常發生,許多染色體異常是會致命的,有些則沒有或僅有極少的作用。據估計,目前總妊娠數中有多達30%是以自然流產結束,而在這當中,大約半數流掉的胎兒有某種形式的染色體畸變。染色體發生變化的後果可能遠不如多出或少掉一整條染色體來得可怕,這些變化或許是一條染色體上有數個區段重新排列,九_九_藏_書或有一部分轉移到另一條染色體上。如果這造成遺傳物質凈增加或凈損失,如同多出或少掉一整條染色體的情況,這種不平衡的結果通常是有害的。不幸的是,對胎兒染色體進行一般的細胞分析,只能偵查到重大的失衡情況,但有時即使是較不重大的失衡情況也會造成非常嚴重的後果。
由於發育中的胎兒必須夠大,才能安全地從其身上取出組織樣本,因此這類診斷不能在懷孕前期進行。一般而言,孕婦是在懷孕第15周到第18周時接受羊膜穿刺術(amniocentesis),這種檢測法是取出一些羊水來進行化驗(羊水中自然含有胎兒的細胞)。另一種可以在懷孕第10周就進行的檢測法,則是收集絨毛膜絨毛(chorionic villus)的細胞,絨毛膜絨毛是胎盤連結子宮壁的部分,但這種方法比較不可靠。由於兩種方法都略具危險性(羊膜穿刺術造成流產的幾率是1/100,而絨毛膜絨毛採樣術造成流產的幾率是2/100),因此一般不建議比較年輕的孕婦做這些檢測。事實上,她們的胎兒有遺傳缺陷的幾率比這些檢查術傷害胎兒的幾率還低。取出的胎兒細胞以前一度必須在培養皿中培養,然後才能進行染色體分析。現在已經有了熒光原位雜交技術(fluorescence in situ hybridization, FISH),診斷速度更快。這種方法是把一個小熒光分子貼到第21號染色體特有的一段DNA序列上,再注入樣本中,接著它會連結至胎兒第21號染色體的DNA。如果細胞核上有兩個熒光點,這個胎兒是正常的;如果有三個熒光點,這個胎兒則有唐氏症。
篩檢纖維囊泡症的困難之一在於技術問題,與造成這種病的潛在缺陷的變異有關。在纖維囊泡症病例當中,大約有70%是由一種特定形式的突變所引起,稱為△F508缺失(deletion),被刪除的三鹼基是CTT。如果其餘30%只是由少數幾種其他形式的突變所引起的,那麼針對纖維囊泡症帶因者進行全面性的族群篩檢(population screening),便不至於不切實際。問題就在於大多數會造成纖維囊泡症的突變只發生在單一家族,而目前為止已發現超過1000種導致纖維囊泡症的不同突變。這在族群篩檢上有何意義?實際上,任何篩檢最多能找出25種不同的突變,但這25種最常見的形式仍僅占所有病例的85%。因此,大約每6個突變當中,就有1個不會篩檢出來;以診斷來說,這樣的命中率並不高。現在,假設有一對夫婦在接受極不完善的篩檢后,兩人都證明沒有纖維囊泡症突變。但我們實在沒法跟他們保證,他們的子女絕對不會得到這個病。人們難免要問:既然如此,那我們幹嘛要花300美元做一個無法獲得結論的篩檢?
南西·魏克斯勒來自亨廷頓氏症家族,安妮則來自乳癌家族,她們倆都屬於可以利用新篩檢法來了解其遺傳命運的新一代。隨著我們對比較常見的成人疾病,例如糖尿病和心臟病等的遺傳原因更加了解后,生物科學的預測力量將會愈來愈強,讓我們得知攸關我們每個人的遺傳命運。
遺憾的是,在今日,婦女得知自己的胎兒有唐氏症后仍然只有兩個選擇:成為唐氏症寶寶的母親或流產。由於病情輕重程度不一,要作這種痛苦決定更加困難。唐氏症患者都具有唐恩發現的臉部特徵(臉平而寬、鼻子小、眼皮窄而斜),但他們的智商差異很大,從20到85不等(亦即從重度遲緩到低度正常)。他們特別容易罹患多種疾病,包括心臟病(造成15%的唐氏症兒童在一歲內死亡)、腸胃道異常、白血病,而隨著年齡漸增,也有可能罹患白內障和阿爾茨海默氏症,但是,他們也可能只有相對很少的健康問題。如今照顧的環境日益改善,而我們對多出的染色體所造成的風險也更加了解,唐氏症患者的預期壽命已經大幅增加,有50%的患者可以活到50多歲。儘管唐氏症患者在一生中,上醫院會變得有如家常便飯,許多人會覺得這令人沮喪,但是一般而言,唐氏症患者仍可以享受人生,也可以為許多家庭帶來歡樂。在有唐氏症患者的家庭中,父母承受的壓力可能比較大,因為他們必須調整生活,照顧這個有特殊醫療需求的孩子,同時知道他們可能在許多方面永遠不會長大。
隨著我們對自身的分子構造日益了解,在這場基因賭博中不幸抽到下下籤的人是否勢必得付出某種代價?我們憑什麼認為這類歧視待遇僅限於保險業者?我的DNA可能顯示我有罹患心臟病、中風、酗酒或憂鬱症的風險,這類信息會不會讓僱主在僱用我以前三思?
最後韋伯和柯林斯還是決定,安妮的困境比遵守規則重要。他們通知安妮,她得病的幾率較低,於是安妮在鬆了一大口氣后取消了手術。但是在把他們的發現告訴這個家族的成員之一后,這些研究人員覺得有義務讓想知道的人享有相同的好處;於是韋伯和柯林斯特別成立了一個乳癌遺傳諮詢計劃。這個家族中有一位證實患病幾率並不特別高的成員,已經在五年前動了預防性雙側乳|房切除術。她對這個遲來的診斷看得很開,她的想法是,動手術讓她的心靈平靜了五年。但是如果她的檢測結果是有突變,那麼當年這個激烈做法可以帶給她的,可能不僅是心靈上的平靜而已。先前有許多年,醫生會建議做預防性乳|房切除術,即便沒有任何手術可以完全清除乳|房組織,當時也沒有確鑿的數據證明這個方法拯救了一些人的性命。然而現在已經有證據證明,這種極端的做法的確降低了乳癌高危婦女的死亡率。以一項調查為例,在動了這手術的639人中,只有2人真的死於乳癌,而不是統計數字預期的20到40人。同樣地,在40歲前(但在已經不再生育之後)切除卵巢,可降低卵巢癌和乳癌的幾率。遺傳分析賦予了婦女作決定的能力,而這真的可以造成生死差別。
今日我們在處理遺傳疾病時,能做的已經比20年前多得多;例如唐氏症和纖維嚢泡症患者的預期壽命不斷增長,就是醫學進展的證明。但是目前我們最強大的武器仍是早期的診斷。至於要不要接受篩檢,最好是留給個人或父母來抉擇,因為他們必須直接承受這些基因信息所帶來的負擔。就產前診斷而言,則應由孕婦來作決定。這並不是說他人不用參与,而是說最終的選擇權應該歸於孕婦,不僅因為她是胎兒的母親,也因為無論我們喜不喜歡,現今的世界仍是由婦女來承擔日常照顧兒童的責任。然而,無論作決定的各種考慮為何,對我來說有一件事是很明確的:長久以來,遺傳疾病已經給無數家庭帶來無法想像的悲慘後果,凱蘿·卡利飽受亨廷頓氏症折磨的家庭就是一例。篩檢可以藉由預防遺傳疾病的發生來減少這類不幸。在發展出篩檢方法后,不把它們告知那些可能想採用的人是不合乎良知的,而不讓所有人都能採用它們,更是令人無法原諒的做法。
從這類問題看來,在赫胥黎想像的25世紀來臨前,他筆下的《美麗新世界》(Brave New World)就已然堂堂登場了。DNA已經深入21世紀的生活,就像一個無法再關回瓶中的精靈。至於我們要如何運用它,決定權則在於我們這個民主社會。不幸的是,在民主社會中,立法的速度似乎總是跟不上實際的需求:一般而言,通常要有數起車禍發生后,某個危險的十字路口才會裝上紅綠燈。可能要到有一些極端不公平的可怕事件發生,有人因自己的基因組而成為受害者之後,才會有推動適當法律通過的動力。這應該是什麼樣的法律昵?「基因隱私」應該是基本準則,但不一定是最終目標。基因隱私應該與社會的其他優先事項達成平衡,尤其是在對抗疾病上。在這方面要有所進展,醫學研究人員能否盡量取用大眾的基因資料顯得日益重要。立法不應阻擋我們充分開發DNA的諸多潛力,包括減輕人類痛苦、告訴人們有關其本身及祖先之事,或協助找出罪犯等等,而且它至少要能確保,所有人民不會因其基因而被剝奪民權或人權。
有時照顧患者比本身患病更令人難以忍受。喬治亞州漢普頓市的凱蘿·卡利(Carol Carr)一直照看著她丈夫霍伊特(Hoyt),霍伊特在30多歲時罹患亨廷頓氏症。他姐姐羅絲琳已經因這種病過世,他哥哥則在獲知診斷結果后立即自殺。凱蘿辭掉工作,成為霍伊特的全職護士,照顧了20年。在霍伊特被診斷出來前,他們已育有三子,而當他在1995年病逝時,凱蘿已經開始照顧兩個年長的兒子藍帝和安迪,就跟照顧她丈夫一樣,喂他們吃飯、替他們洗澡、給他們吃藥、幫他們上廁所。小兒子詹姆士也很快顯現癥狀,絕望的凱蘿只好把兩個大兒子送到私人療養院。2002年6月8日,她在療養院槍殺了他們倆。根據《紐約時報》的報道,詹姆士說,亨廷頓氏症早在他心碎的母親扣下扳機之前,就已經殺死了他的兩個哥哥。
結果,原先大家期望的療法最後證明是一場空。戴維仍以「泡泡」為家,而他也引起全國的注意。美國太空總署嘗試幫助他,為他製造了一個活動型生物支持隔離系統(Mobile Biologistical Isolation System),基本上,這是一套讓戴維可以自由地到「泡泡」外探險的太空裝。但是,就算太空裝也只是另一種形式的「泡泡」。
儘管遺傳篩檢在許多方面的確有用,但是總會引起爭議。小兒科醫師海格曼(Randi Hagerman)在丹佛兒童醫院服務時,決定對丹佛特殊教育班的兒童進行X染色體脆弱症的DNA檢測。她的理由很簡單:如果能找出患有這種病的兒童,就可以給予這些因病導致學習能力受損的兒童更好的照顧,依照他們的特殊需求來規劃課程。結果在439個受檢學生中,發現5人有X染色體脆弱症。(在荷蘭一個規模更大的學校調查,1531名學生當中,找到11個先前沒有診斷出的X染色體脆弱症病例。)
這種基於嚴酷現實考慮的邏輯也適用於較小的國家,在這類國家,能夠承受政策錯誤的資金更小。在以色列,一個對14334名婦女所作的試驗性篩檢研究發現,其中207人有前突變。在孕婦要求下所做的產前診斷,找出5個CGG重複情況已經擴增的胎兒。這些胎兒的命運當然是由孕婦來選擇:自由社會不應要求婦女墮掉有遺傳疾病的胎兒,也不應要求她將這樣的胎兒生下。然而並非每位婦女都有扶養殘障子女的準備,也不是每位婦女都會因為孩子未來的生活質量已可預見,而願意終止懷孕。不過,無論個人的選擇是什麼,不變的事實是:篩檢絕對會降低這種疾病的發生率,而這對社會毫無疑問是件好事。
並非所有的遺傳疾病都會因為醫學無能為力而成為悲劇,最好的實例或許要算是促使一些食品(特別是汽水等不含酒精的飲料)包裝上出現細字警語「含苯丙氨酸」的疾病。苯丙氨酸是一種氨基酸,但是患有遺傳疾病苯丙酮酸尿症(phenylketonuria, PKU)的人無法處理這種氨基酸。
隨著CGG重複數逐代增加,病情也愈發嚴重,而疾病徵候的開始年齡也逐漸降低。X染色體脆弱症家族裡最近一代的子孫,CGG重複數最多,而且通常發病年齡較早,病情也比他們的上一代嚴重。因此遺傳學家可以找出攜帶「前突變」(premutation)的人,這些人的CGG重複數雖然尚未多到會在自己這一代引發疾病,但是從下一代的重複數有可能擴增的情況來看,已經足以使其後的世代罹患X染色體脆弱症。我們目前尚未確定這些突變基因所製造的蛋白質有哪些作用,但是它們似乎會結合至突觸(synapses,神經細胞之間的接合區)中的信使RNA分子上。